|
STAMP Help |
| Name | Description and Recognition Rules | Example |
|---|---|---|
| TRANSFAC | STAMP recognizes TRANSFAC format matrices by the presence of "DE", "NA" or "P0" tags as the first words on a line. If the "DE" or "NA" tags are present, the motif name is taken as the next word on the line.
The frequency matrix is read as the first 5 or 6-column line after the DE, NA or P0 line. All other TRANSFAC tags (AC, ID, BF, XX, etc.) before or after the matrix are ignored. The format of the matrix is: Column1: Matrix position Column2: A frequency Column3: C frequency Column4: G frequency Column5: T frequency Column6: Optional consensus letter The recording of the frequency matrix stops at the next motif-format tag encountered or when the number of columns in a line drops below 5. |
NA Mync XX DE Mync XX P0 A C G T 01 0 31 0 0 C 02 29 0 0 2 A 03 0 30 0 1 C 04 2 1 28 0 G 05 0 3 0 28 T 06 0 0 31 0 G XX |
| TRANSFAC-like | This format is a simplification of the TRANSFAC format described above. The format of the matrix remains the same, but each matrix is directly preceded by a line beginning with a "DE" tag and followed by the motif name. This format is used by the SOMBRERO motif-finder. |
DE Mync 01 0 31 0 0 C 02 29 0 0 2 A 03 0 30 0 1 C 04 2 1 28 0 G 05 0 3 0 28 T 06 0 0 31 0 G XX |
| Raw PSSM | As with the TRANSFAC formats, this format represents motifs as Lx4 matrices. In the "Raw PSSM" format, the matrix is directly preceded by a line beginning with the > character. The name of the motif is taken as the next word after the > character. The matrix itself consists of a set of 4-column lines in the order A C G T. |
>Mync 0 31 0 0 29 0 0 2 0 30 0 1 2 1 28 0 0 3 0 28 0 0 31 0 |
| Jaspar/Consite | This format is used by the Jaspar database and Consite program. It is the only 4xL matrix format currently recognized by STAMP. The Jaspar motif must be preceded by a >-containing line which also contains the motif name. In addition, the four rows of the matrix must begin with the DNA letter represented by the frequencies in that row (i.e. A C G T). Square brackets "[]" may be present in the motif (as they are on the Jaspar webpages), but they will be ignored and are not required. |
> Mycn A [ 0 29 0 2 0 0 ] C [31 0 30 1 3 0 ] G [ 0 0 0 28 0 31] T [ 0 2 1 0 28 0 ] |
| MEME output | The MEME motif format is recognized by the presence of a line beginning with "letter-probability". This line must be immediately followed by a 4-column frequency matrix in the order A C G T. |
------------------------ Motif 2 position-specific probability matrix ------------------------ letter-probability matrix: alength= 4 w= 6 nsites= 31 0 31 0 0 29 0 0 2 0 30 0 1 2 1 28 0 0 3 0 28 0 0 31 0 |
| Consensus sequence | STAMP converts consensus sequences into probability matrices using literal IUPAC rules. The definition of degenerate characters are as follows: M = A or C (i.e. A=0.5 C=0.5 G=0 T=0) R = A or G W = A or T S = C or G Y = C or T K = G or T V = not T (i.e. A=1/3 C=1/3 G=1/3 T=0) H = not G D = not C B = not A N = A=C=G=T=0.25 |
>RUNX1 NNWCTYGGTY |
| Sequence Alignment | STAMP allows the input of sequence alignments, from which a frequency matrix is constructed. Each input alignment must be preceded by a >-containing line that also has a name for the alignment. All sequences in the alignment must be the same length as each other. The aligned sequences may contain gap characters ("-" or ".") or consensus sequence letters. |
>Align1 AACACGTGGC GCCACGT-CC CGCATGTGCA A--ACGTGTT NACWCGTGCC |
| XMS | XMS is an XML format for describing regulatory motifs and PSSMs. This format was defined by Thomas Down, and used in the NestedMICA and MotifExplorer programs. | XMS sample |
| Name | Description and Recognition Rules | Example |
|---|---|---|
| SOMBRERO | The SOMBRERO output files are too large to be uploaded to STAMP. However, you can use this Perl script to convert the top X number of motifs found by SOMBRERO into an acceptable TRANSFAC-like format input file. | Example of output from the TopX.pl script applied to the results of a SOMBRERO analysis. |
| BioProspector or CompareProspector | Bioprospector output files are recognized if the first 5 lines of the file contain the string "BioProspector" and two lines later a string of "*******" appears. BioProspector motifs begin with a line starting with a "Blk1" or "Blk2" tag and the matrix consists of a set of 9-column lines, of which columns 2-5 are A C G T frequency counts. | BioProspector output. |
| MDscan | MDscan's motifs are returned in the same format as BioProspectors. However, MDscan uses two different file formats (one from the command line version and one in the emailed results from the online version). The command-line version output files are recognized via a series of unique keywords in the first 5 lines of the file ("Pm", "Mtf" and "Final Motif"). The email version is recognized via the tag "MDscan Search Result" followed two lines later by a string of "*******". | MDscan output. |
| AlignACE | AlignACE output files (from the command-line version of AlignACE) contain the string "AlignACE" as the first word in the file. The motifs are represented as a type of sequence alignment, which STAMP parses and converts into frequency matrices. | AlignACE output. |
| MEME (text file output) | The second or third line in MEME output files begin with the word "MEME". The "position-specific probability matrix" parts of the file are scanned for and parsed as described as above for individual MEME-format motifs. | MEME output. |
| Weeder | Weeder output files always contain the line �**** MY ADVICE ****� followed three lines later by �*** Interesting motifs (highest-ranking) seem to be�. Motifs are marked by the string �Frequency Matrix� and consist of a set of 9-column lines (beginning 3 lines after the marker), of which columns 2-5 are A C G T frequency counts. | Weeder output. |
| MotifSampler | STAMP recognizes MotifSampler matrix files (these are the .mtrx files emailed from the online version). The files always begin with the string "#INCLUSive Motif Model". PSSMs are represented as 4-column frequency matrices preceded by some description lines. | MotifSampler output. |
| YMF | STAMP recognizes both YMF command-line and web program output. The formats begin (respectively) with the lines "The best X candidates in category Y are :" or "Motif Count Zscore". YMF motifs are given one-per-line in consensus sequence format. | YMF output. |
| ANN-Spec (command-line only) | ANN-Spec command line output files begin with the line "SQI SEQUENCE_INFORMATION:". The predicted motifs (in 4xL matrix format) are prefaced with "ALR" tags. | ANN-Spec output. |
| Consensus (command-line only) | Consensus output files contain the string "THE LIST OF MATRICES FROM FINAL CYCLE--" Motifs are in 4xL format (JASPAR-like). | Consensus output. |
| Improbizer | STAMP recognizes Improbizer HTML output (as opposed to the motif file output from the command-line version of the program). Improbizer output begins with the text "Improbizer Results", and the motifs are in 4xL format, where the frequencies begin with lowercase nucleotide tags. | Improbizer output. |
| Co-Bind | Co-Bind output file's first line contain the string "# reading predefined alphabet from file". The second line contains the string "# ***** sequence information from sequence set". The matrices are in 4xL format, coming after the "ALIGNMENT_MATRIX" tag. | Co-Bind output. |
| DME or CREAD | DME and CREAD use a format that is similar to the TRANSFAC format described above. | CREAD sample |
| NestedMICA | NestedMICA's output format is the XMS (XML-like) format described above. | XMS sample |
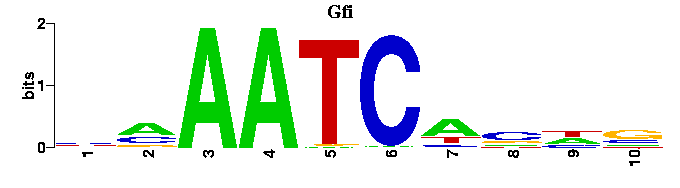 into this:
into this:
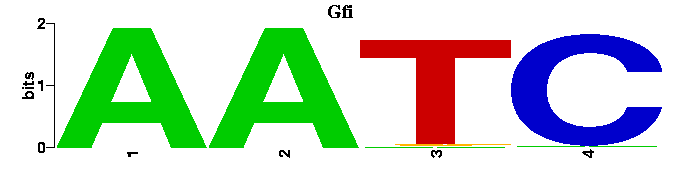
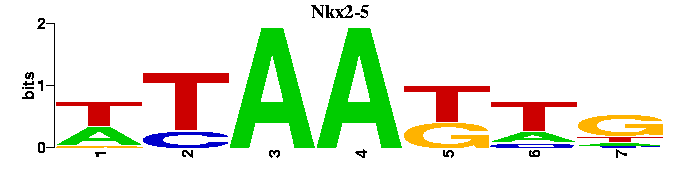 into this:
into this:

Firstly, a multiple alignment of the input motifs is provided. While the multiple alignment was carried out on the motif matrices themselves, the multiple alignment is represented using consensus sequences (for implementation simplicity).
The consensus alphabet used here is not the literal IUPAC consensus sequence rules (using this usually leads to consensus sequences that are degenerate beyond recognition), but rather using the following probability thresholds:
A/C/G/T is used if the appropriate single base frequency is >0.6
M/R/W/S/Y/K is used if the sum of the appropriate two bases is >0.8
N is used otherwise.
A "familial binding profile" based on the final multiple alignment is also provided as a sequence logo and as a TRANSFAC-format frequency matrix.
A tree showing the similarity between the input motifs is shown next. This tree should be useful when trying to elucidate the relationships between motifs of transcription factors from the same structural class or when trying to find degeneracy in the results of motif-finders. Next to the tree figure, the input motifs are displayed as sequence logos in the same order as the appear on the tree. Alongside the input motif logos, the best match in the chosen motif database is also shown as a sequence logo.
Finally, for each input motif, a number of the top database matches (as decided by the user) are shown in more detail. An alignment between the input motif and the match is shown as a consensus sequence. The p-value of the alignment is also shown. The p-values are calculated using the methods described by Sandelin & Wasserman. While this method aims to be independent of the motif lengths, it is not perfectly so. Therefore, it is possible to get a high score even if the motifs do not seem to be all that similar, especially if one of the motifs is large. For this reason, the p-value should not be taken as an accurate measure of the probability of two motifs being identical, but rather as a relative measure of similarity.